Spatial epigenome–transcriptome co-profiling of mammalian tissues
The article titled “Spatial Epigenome–Transcriptome Co-Profiling of Mammalian Tissues” published in Nature, presents groundbreaking methodologies that integrate spatial epigenomics and transcriptomics to co-profile chromatin accessibility and gene expression in the same tissue sections. This dual-layered approach provides a more detailed understanding of gene regulation within the tissue architecture, enabling the identification of spatial relationships between epigenetic modifications and transcriptional activities at near-single-cell resolution.
Context and Motivation
Emerging technologies in spatial biology have expanded the capability to analyze cellular states within their native tissue environments, offering insights into how cells function and interact. However, existing methods have been limited by their focus on a single layer of omics information—either epigenomic or transcriptomic data—thereby missing the critical connections between these layers. The methods developed in this study address this gap by enabling the simultaneous capture of spatial epigenome and transcriptome data, specifically through two techniques: spatial ATAC–RNA-seq and spatial CUT&Tag–RNA-seq.
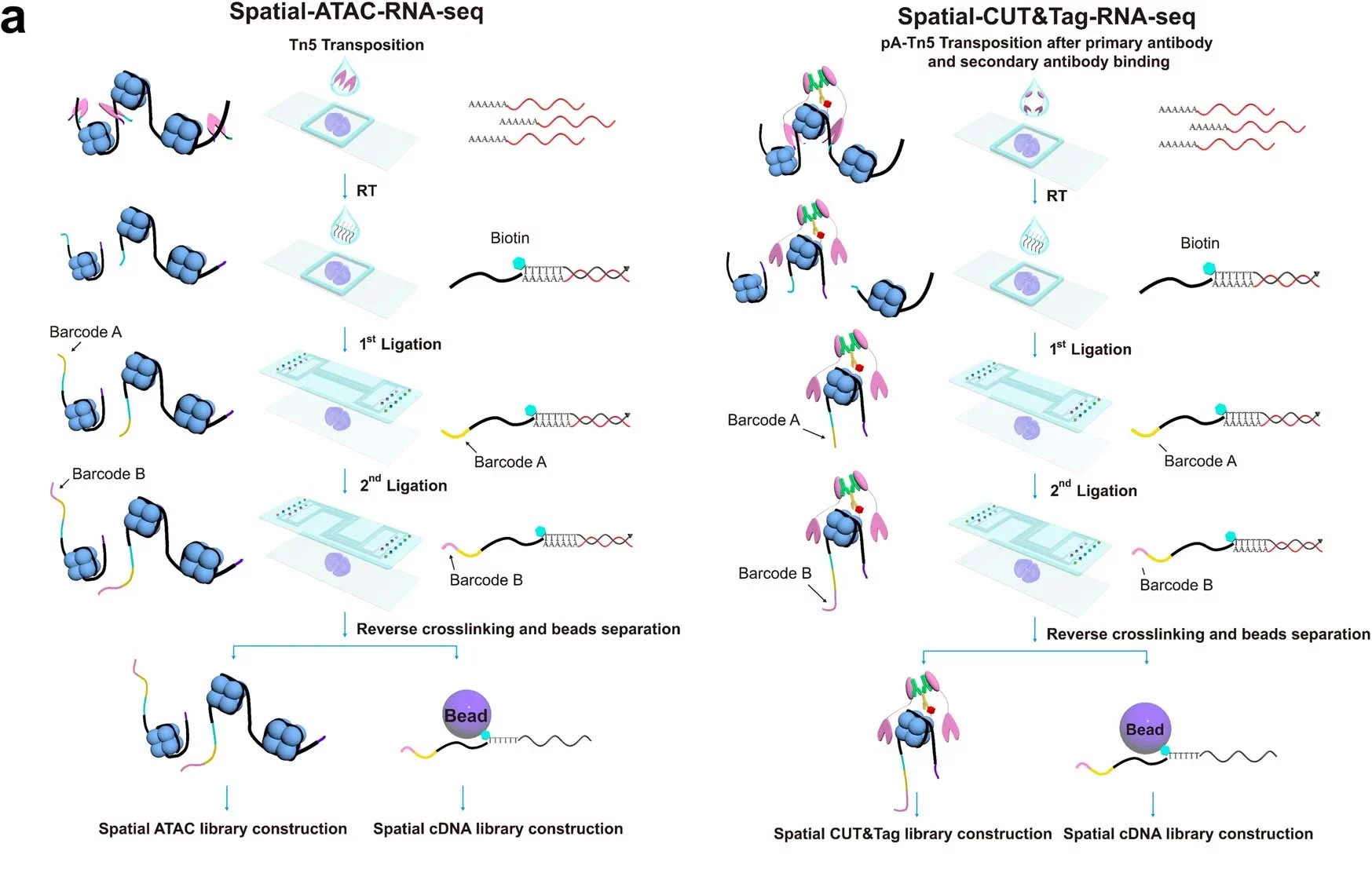
Methodology
Spatial ATAC–RNA-seq
This method allows for the co-sequencing of chromatin accessibility and mRNA expression from the same tissue section. The process begins with the fixation of tissue sections followed by transposition using Tn5 loaded with DNA adaptors. A crucial innovation in this technique is the use of a microfluidic system to spatially barcode the tissue sections. The PDMS microfluidic chip contains a grid of microchannels that introduce spatial barcodes in two perpendicular directions. This creates a unique identifier for each tissue region, forming a 2D grid of spatially barcoded tissue pixels. This setup enables precise mapping of chromatin accessibility and gene expression across different tissue types, such as embryonic mouse brain and adult human brain hippocampus, at near-single-cell resolution.
Spatial CUT&Tag–RNA-seq
This technique extends the spatial co-profiling capabilities to include histone modifications (chemical changes to the proteins around which DNA is wound, affecting gene expression by altering the accessibility of the DNA to the cellular machinery) : H3K27me3, H3K27ac, H3K4me3, along with mRNA expression. Here, a similar microfluidic system is used to apply spatial barcodes to the tissue sections after the introduction of specific antibodies against histone marks. The use of the microfluidic device ensures that these barcodes are applied consistently across the tissue, allowing for the simultaneous mapping of histone modifications and gene expression. This method, like spatial ATAC–RNA-seq, leverages the microfluidic system to maintain high resolution and accuracy in spatial co-profiling.
Key Results
Mouse Brain Development
The application of spatial ATAC–RNA-seq to embryonic and juvenile mouse brains revealed that chromatin accessibility does not always align perfectly with transcriptional profiles. For example, certain genes like Sox2 and Pax6, which are critical in neurogenesis, displayed high chromatin accessibility in specific brain regions but showed varied levels of mRNA expression. This discrepancy suggests epigenetic priming, where chromatin is prepared for future transcriptional activity.
Human Brain Analysis
In adult human hippocampal tissue, spatial co-profiling uncovered significant insights into the gene regulation of cognitive functions. Notably, genes associated with granule cell layer neurons, such as PROX1 and BCL11B, showed distinct patterns of chromatin accessibility and gene expression, shedding light on the spatial regulation of neurogenesis and brain function.
Epigenetic Regulation Insights
The study highlighted how spatial epigenetic modifications correlate with gene expression at different stages of tissue development. For instance, the loss of chromatin accessibility at the Pax6 locus was observed to precede the downregulation of its corresponding RNA during the transition from neural progenitor cells to neurons. Conversely, genes involved in later stages of neuronal development showed increased expression despite already accessible chromatin, indicating the presence of epigenetic priming.
Implications and Future Directions
The technologies presented in this study are poised to transform the field of spatial biology by providing a comprehensive toolkit for dissecting the mechanisms of gene regulation in various tissue contexts. These methods can be applied to a broad range of biological and biomedical research areas, including developmental biology, neuroscience, and disease pathology. The ability to map epigenomic and transcriptomic layers simultaneously in spatial context offers unprecedented opportunities to explore how local tissue environments influence cellular behavior and how these interactions may go awry in disease states.
Moreover, the methodologies could be expanded to include additional layers of omics data, such as proteomics, further enriching the multidimensional understanding of cellular states and interactions. The integration of spatial omics data could also lead to the development of new computational tools for the analysis and visualization of complex datasets, enabling more precise and predictive models of tissue biology.
Conclusion
This study introduces powerful new tools for the spatial co-profiling of the epigenome and transcriptome, offering detailed insights into gene regulation within tissue architectures. These technologies bridge the gap between different layers of omics data, providing a more complete picture of cellular dynamics and gene regulation in both normal development and disease. The methodologies developed by Zhang, Deng, and colleagues represent a significant advance in spatial biology, with broad implications for future research in various fields.
Reference
The research was conducted by a team from various institutions, including the Department of Biomedical Engineering and the Yale Stem Cell Center and Yale Cancer Center. The full study is available in Nature (2023), DOI : 10.1038/s41586-023-05795-1.

 Job
Job Collaborations
Collaborations Customer
Customer Other
Other